Advance/NeuralMD is a software for molecular dynamics calculations based on a Neural Network force field.
Using the results from first-principles calculations output by Quantum ESPRESSO as
training data, you can create a molecular force field. This force field
will then be employed to perform molecular dynamics simulations using LAMMPS. It incorporates various state-of-the-art technologies, such as automatic force field generation using self-learning hybrid Monte Carlo methods, acceleration on GPUs to enable calculations for systems with up to 100,000 atoms, and a pre-existing database of force fields.
Features of Advance/NeuralMD
1) Force Field Definition Based on 3G-HDNNP
The Neural Network force field implemented in Advance/NeuralMD is based on the High-Dimensional Neural Network Potential (HDNNP).
It also includes the utilization of a third-generation algorithm
(3G-HDNNP) that accounts for long-range Coulomb interactions based on
the charges of individual atoms. Furthermore, by combining our
proprietary Δ-NNP method and techniques that utilize the average
of multiple Neural Network models, we can create a stable force field
with a relatively small amount of training data.
There are examples of applying these methods to calculate the
lithium-ion conductivity of solid electrolytes and analyze the melting
points of nuclear fuel materials.
Case 1: Conductivity Calculation of Lithium Ion Conductor LGPS by Δ-NNP Method
Case 2: Melting Point Analysis of Nuclear Fuel Materials Using Multiple Neural Network Models
|
|
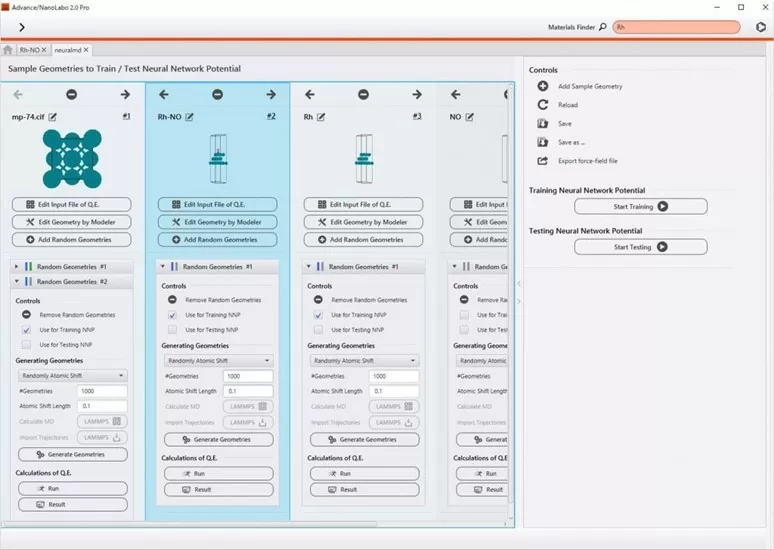
(2) Operations from Advance/NanoLabo
From
creating training data, learning neural networks, and generating force
fields to conducting molecular dynamics simulations, all processes can
be operated from the Advance/NanoLabo interface. In the Grand Project
(see the right figure), you can manage a large number of training data
and proceed with your work. Furthermore, in the process of creating
training data, it is also possible to distribute first-principles
calculations to multiple computational resources such as calculation
servers and the cloud.
|
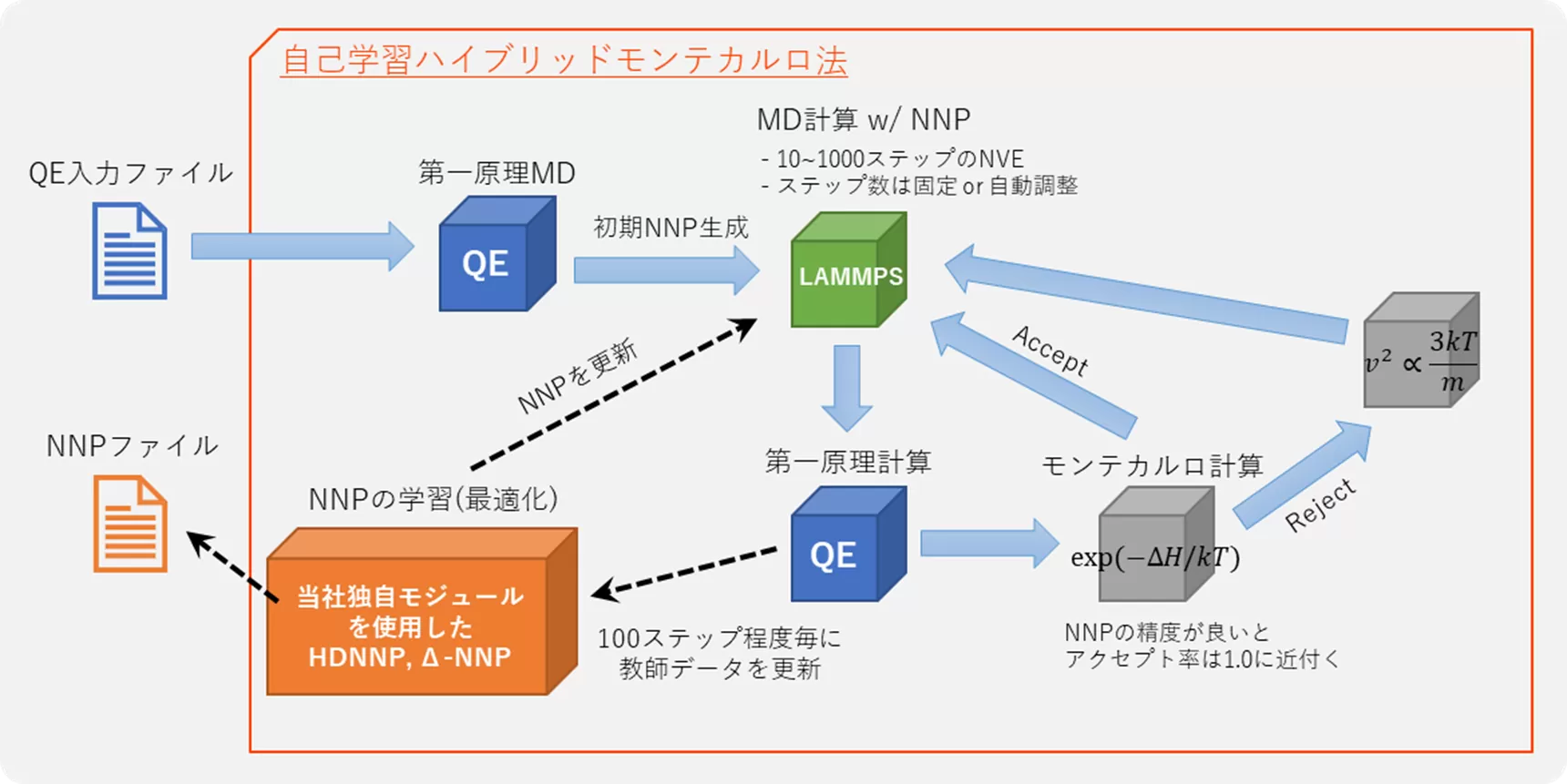
(3) Self-Learning Hybrid Monte Carlo Method
Self-learning hybrid Monte Carlo method
is an algorithm for first-principles Monte Carlo simulations developed
by the Japan Atomic Energy Agency. By utilizing trajectories from
molecular dynamics simulations with Neural Network force fields as
proposed structures in the Monte Carlo method, it is possible to
guarantee accuracy in Monte Carlo calculations comparable to first
principles, while achieving efficient sampling of structures.
Simultaneously with the execution of Monte Carlo calculations, the
learning of the Neural Network force field is also conducted in
parallel, using the results of first-principles calculations performed
for each structure. As a result, when this method is executed, a Neural
Network force field specialized for the target system is automatically
generated.
|
|
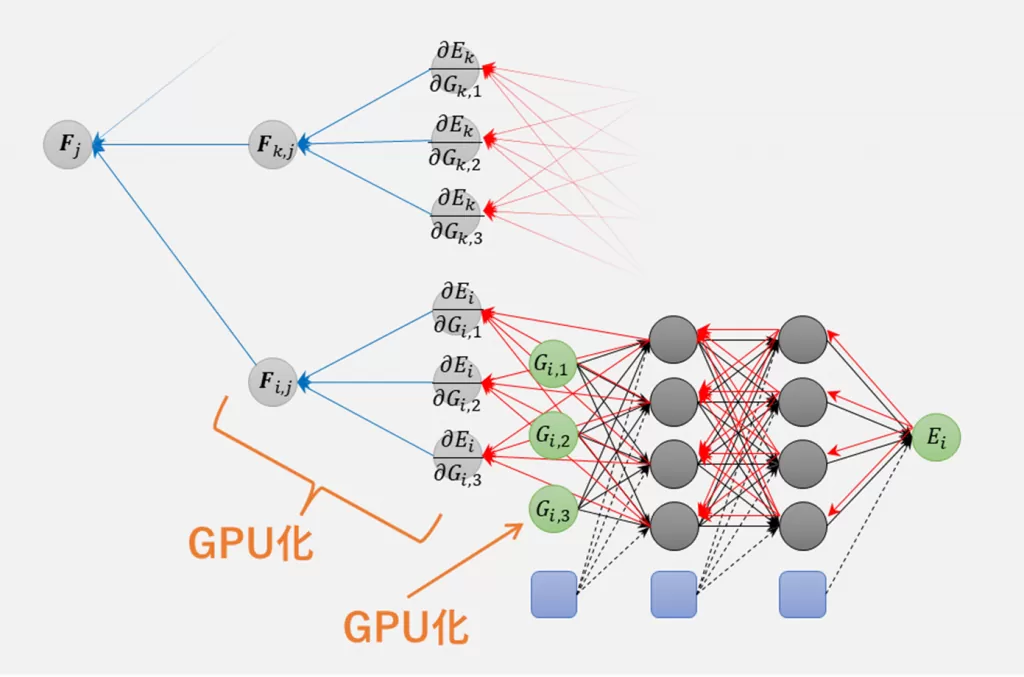
4) Acceleration with GPU
Advance/NeuralMD
Pro supports neural network training and molecular dynamics simulations
on GPUs. It can also be used in conjunction with MPI parallelization
and is compatible with machines equipped with multiple GPUs and/or
multiple machine nodes with GPUs. It is designed to maintain high
utilization of both GPU and CPU by launching 2 to 4 MPI processes per
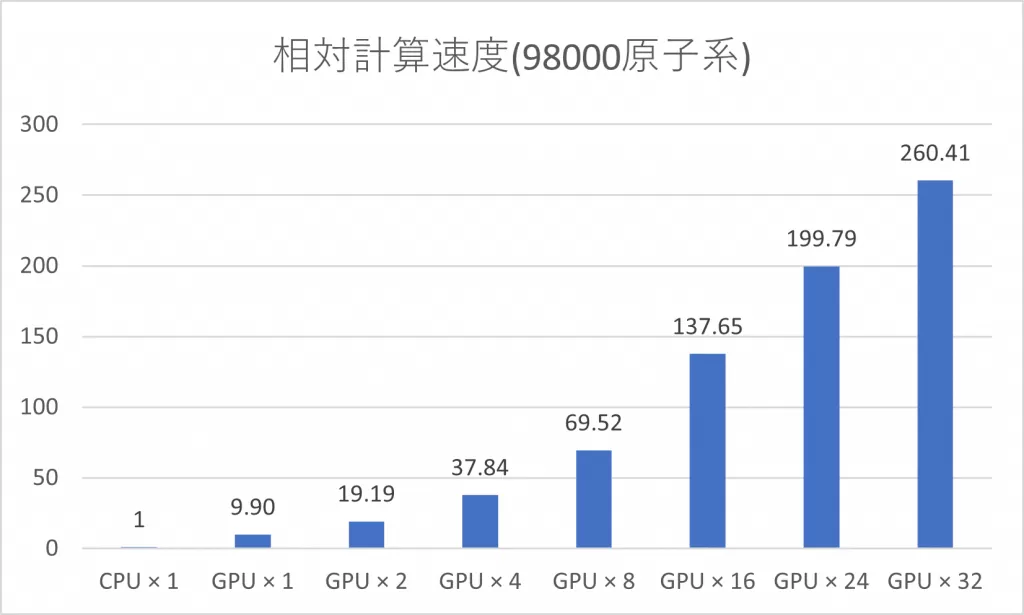
GPU device. We have GPU-accelerated the computationally expensive
symmetric function and force calculations (bottom left). By utilizing
32 GPUs, we have successfully achieved approximately 260 times faster
molecular dynamics simulations for systems with around 100,000 atoms
(using the computing environment provided by HPC systems).
|
 |
 |
| Details of Advance/NanoLabo |
|
|